Clinical
Ritalin Causes Cancer?
An eye-opening study from some Texans.
18 kids, newly diagnosed with ADHD, started the study, only 12 finished. They showed up on day 1, and blood was taken. The kids were then given Ritalin (methylphenidate) 20-54mg/d, as part of ordinary treatment, for three weeks. At the end of three weeks, another blood sample was taken. The bloods were evaluated for cytogenetic abnormalities.
In every single case, the frequency of chromosomal aberrations, sister chromatid exchanges (SCE), micronuclei, and nucleoplasmic bridges were all dramatically higher than at baseline. Not a little higher-- massively higher.

The authors had, in their introduction, summarized the absence of substantial evidence (or actually even studies) for carcinogenicity or mutagenicity, except one long term (2 year) high dose study in rodents-- it gave them hepatocellular carcinoma. But there has been nothing done in humans.
There are some problems with the study, beyond the obvious small sample size.
First, there's no control group. The assumption is that the only new factor over the three months of the study was the taking of Ritalin, so that is the likely culprit. Of course, it is certainly possible that something else occurred during those three months that could have caused this effect, such as a new illness, new meds, taking up smoking, etc. In all twelve people. At the same time. Sure, it's possible.
Second, the pretreatment group actually had less sister chromatid exhcanges than are expected on average. In a follow-up letter letter , the authors indicate that the known average frequencies of SCE are actually based on adults, not kids. Do kids have lower frequencies in general? Maybe.
The authors in that same letter also observe that despite the perception that there has not yet been on observed link between Ritalin and carcinogenicity, in fact
"the national toxicology program (NTP)—CERHR expert panel report on the reproductive and developmental toxicity of methylphenidate, indicate that only one study addressed the carcinogenic risk of methylphenidate treatment in humans... conducted by screening pharmacy and medical records, indicated that there was no increase in reports of cancer in a small number of patients taking methylphenidate (only 529 patients)."
I looked up the cytogenetic effects of amphetamines.
One study found methamphetamine exposure correlated to frequency of micronuclei and SCEs in humans (though, in hamsters, this effect was due exclusively to methamphetamine itself, and not its metabolites; and free radical scavengers also reduced this effect).
An old 2 year rodent study found decreases in number of neoplasms when given dl-amphetamine. Another study found a similar reduction, especially in pheochromocytomas, pituitary adenomas, and breast adenomas.
But again, these are rare studies, and this one here is the first done, prospectively, in humans.
What is astounding to me, apart from the obvious, is that no one knows this article. It has not been referenced in any subsequent articles. I can't find one psychiatrist, academic or otherwise, who has even heard this. They all look at me blankly: "Really?"
Yet, simultaneously, psychiatrists live with complete confidence that Ritalin is safe. They've never checked the known information before, of course, so what allows them to be so confident I have no idea; and they certainly don't run Medline once a month "just to keep up with all of science"-- but they're sure of what they know. Not even an empty patronizing nod to "but of course, our knowledge base is expanding..."
The point is not that Ritalin is unsafe. This study could be a load of crap, for all we know. But shouldn't psychiatry have at least heard of this study? What is the mechanism to disseminate this kind of information? How long does it take for something like this to hit the psychiatric press? In other words, given psychiatrists' arrogant confidence, how do they believe they would be informed of new developments? They don't really read psychiatry journals. They certainly aren't going to read cancer journals.
9/5/06 Update Further info suggests this may be a fluke.

 Score: 2 (2 votes cast)
Score: 2 (2 votes cast)
Liver and Medications
Here's how to think about the effect of the liver on drugs:
When you eat a drug, some of it gets bound to protein (albumin) and some circulates freely. Your body uses free, non-protein bound drug.
Most drugs are mostly protein bound-- notable exceptions are lithium, Ritalin, Lexapro (40%) and Effexor (40%). Low albumin-- as could occur in cirrhosis, severe malnutrition, etc-- increase the availability of the free form of the drugs. So low albumin + valium = more valium for you.
Next is volume of distribution (Vd)-- drugs with high Vd will diffuse into fluid spaces. So patients with a lot of edema will end up with lower useful drug (because it diffused into third spaces.) Be aware that diuresis may consequently increase the dose as it returns to circulation.
First pass metabolism is also important. First pass metabolism means that a substantial portion of the drug is metabolized quickly-- or, conversely, won't be metabolized if your liver is damaged. The appropriate doses of medications are based on functioning livers. For example, tricyclics are metabolized by 50% on first pass; which means, in the absence of a liver, you are actually giving twice as much drug as what you think you are prescribing. Zyprexa has 40% first pass. Dilantin, by contrast, has low first pass metabolism, so the dose is about the same.
There are two phases of liver metabolism.
Phase 1 occurs in smooth endoplasmic reticulum: reduction, oxidation, and hydrolysis. All the cytochrome P450 happens in Phase 1.
Phase 2 occurs in periportal region of portal triad: glucoronidation, acetylation, sulfation.
The trick of this is to understand that liver damage (cirrhosis, etc) affects Phase 1, not Phase 2.
This is why Ativan (lorazepam), Serax (oxazepam) and Restoril (temazepam)-- all metabolized primarily by Phase 2-- are favored in drinkers or cirrhotics. Also, renally metabolized or cleared drugs will not be as much affected (for example, Neurontin.)
I hope this was helpful. Please drink responsibly.
Score: 3 (3 votes cast)
Parenting and Personality: MAO-A
Continuing the series:
The authors investigate the interaction between child abuse and MAOA (gene) activity on future antisocial behavior.
FYI: MAOA is a gene on the X chromosome-- so males only have one copy. It makes MAO-A enzyme, which metabolizes serotonin, dopamine, and norepinephrine, so having low MAOA gene activity probably means less MAO-A, and thus more serotonin, norepinephrine, and dopamine. MAO-A is in CNS, liver, and GI tract; MAO-B (metabolizes mostly dopamine and phenethylamines (e.g. amphetamines)) is in CNS and platelets.
Importantly, having been maltreated in childhood predisposed you to becoming antisocial; having the MAO-A deficiency, by itself, did not. This is important: MAO activity has no effect in the absence of child abuse. Having low MAOA activity does not predisose you to violence. The abuse is the primary determinant.
What about the interaction between the environment (abuse) and biology (MAOA)? This interaction is very significant, but how you explain this interaction makes all the difference. Here's the figure:

The easy (and wrong) explanation, the one that jumps out at you, is this: if you were maltreated, having low MAO-A predisposes you to becoming antisocial.
But that's not what the figure shows. What it shows is that having high MAOA mitigated, i.e. lessened, the effect of being abused on future criminality.

What you see is that when MAO-A is high, you are protected against the effects of abuse. When it is low, abuse matters.
It may seem like the distinction between MAO being protective vs. being a risk factor is only semantics, but it isn't. How we define the problem actually generates different problems. "Having low MAOA increases your risk of being antisocial" is a very different social problem than "having high MAOA lessens the effect of child abuse." So if you're a lawyer, don't go concocting a "low MAOA made me do it" defense.
As an aside, it might be helpful if someone could explain how having low MAOA is a risk factor for agression and violence, but taking antidepressants (and MAO inhibitors especially) are supposed to make you less violent?
Score: 4 (4 votes cast)
Parenting and Personality Disorders
A fascinating article that no one will ever actually read: Parenting Behaviors Associated With Risk For Offspring Personality Disorder During Adulthood.
The authors made a (startling) discovery: there are types of parenting behaviors which predispose your kid to growing up personality disordered.
This was a longitudinal study of 592 families, first assessed when the kids were about 5, and then again when they were in their 30s. (More info at their website http://nyspi.org/childcom/)
The results are pretty much what you'd expect:
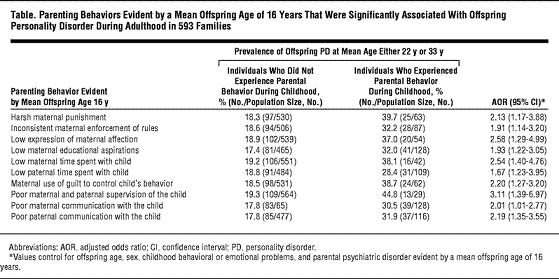
The more of these behaviors the parents exhibited, the more the risk of PD increased. What is interesting is which PD was increased given the number of parental behaviors:
First, overall number of bad parental behaviors:
(antisocial=criminal; avoidant=shy; narcissistic=self-absorbed)

You'll notice that antisocial PD is essentially zero at baseline, and is dramatically sensitive to bad parenting. Contrast this with avoidant PD, which, while also sensitive to the parenting, starts out higher at baseline. In other words, you may be born shy, but not antisocial.
Looking at specific types of bad parenting:
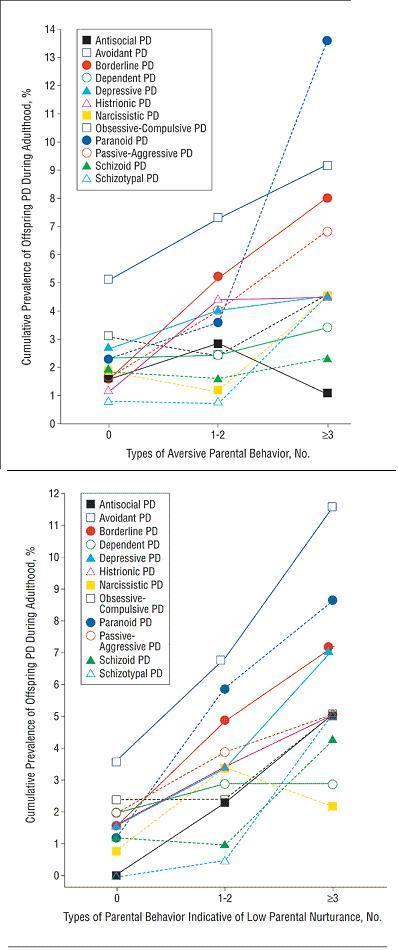
What you'll see in the top figure is that being an aversive parent is a great way of making someone borderline or passive-aggressive, not to mention paranoid. But it doesn't make them antisocial. Hmm.
Meanwhile, having low affection or low nurturing scores increased the risk for antisocial, as well as everything else (but especially avoidant, paranoid, depressive, borderline).
Some covariate caveats: even when parental psychaitric disorders and offspring behavioral problems at age 6 were controlled, bad parenting was still associatd with increased risk of their kids' PD.
Furthermore, the usual association of parental psychiatric disorder leading to child PD could be explained, in fact, 95% due to the bad parenting. Another way of saying this is that 95% of the effect that a parental psychiatric disorder has on causing their kids' personality disorder can be obviated by better parenting. In a similar vein, 35% of the effect of childhood behavioral problems leading to later PD can be similarly reduced by better parenting. In other words, even if you or your kids have a "biological" psychiatric disorder, better parenting skills can darmiatically affect the outcome.
It is not an insignificant fact that only one of the 5 authors was an MD (oddly, he is also a PhD but does not list this in the authorship line.). The nature vs. nurture debate in psychiatry is all but dead.
The longer we delude ourselves that biology controls behavior, and not the other way around, the longer we'll have to live with the same behaviors.
Score: 12 (12 votes cast)
Zyprexa's Weight Gain: Does What You Eat Matter More Than How Much?
The authors of this article have an interesting hypothesis, upon which I speculate wildly. But it is fascinating:
GLUT5, is found primarily in the small intestine (though also in muscle and kidneys.) What's interesting about it is that it transports fructose, which in turn directly stimulates additional GLUT5 mRNA expression. You eat fructose, this increases the expression of GLUT5 in the intestinal villi, which increases the transport of fructose. So the more fructose you eat, the more readily you can absorb it.
Now fructose doesn't stimulate insulin secretion. Since insulin regulates leptin, fructose actually reduces leptin. Fructose increases ghrelin. So you get hungry. Fructose goes to the liver and is metabilized to acyl glycerols, and consequently result in increased triglycerides.
So you have a situation in which Remeron and Zyprexa (and high dose Haldol) cause an increase in GLUT5 expression; if they are also eating fructose (read: high fructose corn syrup) this is causing an additional expression in GLUT5, and hunger, and increased triglycerides... if one wants to conduct a useful experiment, find out if the people who gain the most weight on Zyprexa are those who consume the most high fructose corn syrup (and not just those who eat the most.) In other words, can you gain weight on Zyprexa if you are eating Atkins?
(NB: there are many who want to believe that Zyprexa causes weight gain by increasing leptin; and so fructose and GLUT5 lowering leptin seems confusing. Zyprexa, as shown above, actually decreases leptin, acutely. (And clozaril has either no effect, or minimal lowering.) Letpin only increases with increased fat-- i.e. as a consequence of fat, not as the cause of fat. Those who have found increases in serum leptin do so only after chronic administration, and resultant weight gain (for example, in a study of 13 schizophrenics on Zyprexa who showed a small increase in leptin after 4 weeks-- and after a 2 kg weight gain; or 6 week animal study finding increased fat and leptin. The question, as noted by the authors, is whether the acute hypoleptinemia and hypoglycemia is what triggers hunger and an ultimate increase in fat, leptin, glucose and insulin. )
Score: 2 (2 votes cast)
How Do Antipsychotics Cause Weight Gain?
In order for this post-- and any discussion on antipsychotic induced weight gain-- to make sense, you have to understand one thing: each antipsychotic seems to cause weight gain by a different mechanism, not varying degrees of the same mechanism. Because let me tell you right off the bat: researchers here are far from agreed.
A review of some articles:
In rat pancreatic beta cells, neither clozapine nor haloperidol had any effect on basal insulin release. In the presence of high glucose, haloperidol had no effect on the normal insulin surge, but clozapine inhibited this effect by 40%. How it did this is not clear, as clozapine, in the presence of glucose, completely suppressed electrical activity by hyperpolarizing the membrane potential (i.e. increased K+ conductance.) Haloperidol depolarized (inhibited K+ conductance). Thus, by completely suppressing electrical activity, it should have completely suppressed insulin release-- but it only inhibited 40%. Similarly, haloperidol should have increased insulin release (via depolarization) but it didn't have any effect. We don't know what would have happened if the study had been continued for a year; but note here that the effect on insulin is dependent on the presence or absence of glucose, not the other way around.
Most studies focus on the changes in serum parameters (triglyceride, cholesterol, insulin, etc) and not the mechanism for these changes.
For example, in 112 schizophrenics on meds for 8 weeks, Zyprexa, clozapine, Risperdal, sulpiride all increased insulin and C reactive peptide, as well as insulin resistance; but only clozapine and Zyprexa increased triglycerides and cholesterol, and had a greater impact on insulin, insulin resistance, and C-peptide. What you can't tell is when this happened and what came first: did the insulin go up as a direct effect of the med, and consequently so did cholesterol, or did insulin resistance happen first, etc?
In the first study looking at the drugs' effects on GLUT1-5 mRNA, it was found that Remeron (mirtazapine) increases GLUT4 (muscle/fat) and 5(intestine) mRNA, and Haldol and Zyprexa increase GLUT5. No effect on GLUT1-3. (Contrast with Clozaril and Risperdal, below.)
The authors propose something interesting about Remeron: "Therefore, the increasing effects of mirtazapine on GLUT4 mRNA levels in our study might lead to a decrease in blood glucose levels and to an increase in cellular fat deposition, leading to intermittent or continuous lowering of blood glucose levels with a subsequent increased uptake of carbohydrates and other types of nutrients." In other words, better glucose uptake into cells means more fat inside cells, and less glucose outside cells (hypoglycemia)-- which is a stimulus to eat more.
This is important, so I'll repeat it: the hyperglycemia seen with Zyprexa and Remeron is here proposed to be due to the acute lowering of blood glucose (because of increased transport), and thus an increase in eating and fat deposition, and consequently insulin resistance and hyperglycemia; not a direct affect on glucose metabolism.
(Consistent with Zyprexa's effect on GLUT5 (and not on carbohydrate metabolism, per se), metformin did not prevent weight gain in 40 people on 10mg Zyprexa (all gained 5-6kg in 14 weeks.)In (male C57) mice, over a 6 month period, clozapine, chlorpromazine and quetiapine induced hyperglycemia via effects on glucose transport. Haldol and amisulpiride have little effect on GLUT, and were found not to induce hyperglycemia. Risperdal had a medium effect on hyperglycemia, but at the lower doses.
Using rat pheochromocytoma cells, clozapine and Risperdal both inhibited glucose transport (i.e. GLUT3).
Desmethylclozapine (a metabolite) was an even more potent inhibitor, while clozapine-N-oxide, the other metabolite, had no effect on glucose transport. Clozapine and fluphenazine also inhibited glucose transport in (rat) muscle cells. The drugs block glucose transport in a non-competitive (i.e. allosteric) manner (and tricyclics appear to work in the same way.) What is interesting about this is that different people metabolize clozapine differently, and perhaps those who create more desmethylclozapine get more hyperglycemia than those who make less (and/or more clozapine-N-oxide.
A follow-up study tried to correlate the toxicity of these drugs to cells to their inhibition of glucose transport.
They found that clozapine, desmethylclozapine, Seroquel and fluphenazine were toxic to cells; Risperdal was minimally toxic; and Zyprexa actually promoted cell growth.
Seroquel, Zyprexa and clozapine all inhibited glucose transport about the same amount, and in a dose dependent manner. (Remember: Haldol and sulpiride don't.)
However, if the cells were exposed to drug for a longer time, fluphenazine greatly inhibited glucose uptake, clozapine had no effect, and Zyprexa increased glucose uptake. In other words, the toxic typicals only need a sort exposure to kill a cell, while less toxic atypicals need prolonged exposure. Also, fluphenazine increased GLUT3, and the atypicals had little or no effect (as found above.)
Zyprexa was found not to affect either the basal or the insulin stimulated glucose transport via GLUT1 or 4. (Fun fact: bovine serum albumin (or impurities therein), used to replicate the fact that olanzapine is highly (93%) protein bound, actually increased basal glucose transport, making suspicious all studies previosuly done with BSA.) This contradicts the findins of the Dwyer articles, above, where antipsychotics had inhibitory effects on glucose transport. A possible explanation could be dosing: this study used doses comparable to 20mg, while others used 20x that amount.
Another study, in humans, found that neither Zyprexa nor Risperdal affected acute (3 week) insulin sensitivity. Again, what happens after you get heavy is up for debate.
So what we have here is confusion, but:
1. acute, high dose in vitro studies indicate that typicals>atypicals inhibit glucose transport, but Haldol does not.
1b. Typicals are toxic to cells, atypicals less so, and Zyprexa promotes cell proliferation.
2. Normal dose and human studies show no effect on insulin dependent glucose transport (i.e. GLUT4) but there are effects on small intestine absorption (GLUT5) with Zyprexa and Remeron.
3. Clozapine inhibits insulin release in the presence of glucose, but Haldol doesn't.
4. Acute effects may be different than chronic. i.e. even though antipsychotics may not directly affect insluin resistance or glucose transport, if they make you hungry or increase fat over time, this could result in later insulin resistance, hyperglycemia, etc.
Score: 0 (2 votes cast)
Atypicals and Diabetes: Glucose Transport
Glucose is absorbed through the small intestine into the blood.
All glucose is taken into cells via hexose transporters: this is facilitated diffusion (no ATP). Facilitated diffusion is passive diffusion through a channel made by a transmembrane protein; the proteins are able to open and close this channel. There are many ways channels can be opened/closed: ligand gated (i.e. neurotransmitter receptors), voltage gated (neurons), or, in the case of hydrophilic molecules such as glucose, mechanically gated: the channel is shaped like a closed "V". Glucose goes to the bottom of the V, causes a conformational change and the "V" opens, but closes at the top (makes an upside down "V".) Glucose can pass, and the V recloses. All diffusion is down a concentration gradient.
The hexose transporters are called, randomly, "Glucose transporters 1-5" (GLUT1-5).
GLUT4 is the main transporter in muscle, fat, and the heart. GLUT4 is insulin-sensitive (though it can also be activated by muscle contraction-- go figure.) In the absence of insulin, GLUT4s are stored in cytomplasmic vesicles floating around in the cytosol. If insulin binds to the insulin receptor (an ATP dependent tyrosine kinase receptor-- NOT the GLUT4), a signal cascade is activated that causes the cytpolasmic vesicle to go to and bind to the plasma membrane and lodge the GLUT4 there. The GLUT4 then allows glucose to diffuse through. When insulin disappears, the insulin receptor reconforms, the signal cascade stops, and the GLUT4 pinches off (by clathrin and other contracting proteins in the cell membrane) into a vesicle again (pinocytosis).
Thus, if there is no insulin: even if there is much glucose, there is no signal for the vesicle to go to the plasma membrane and lodge the GLUT4, so there will be no transport of glucose into the cell; so glucose stays high in the blood. Thus we have Type 1 diabetes.
Insulin also stimulates the creation of glycogen in the liver and muscle. [Insulin activates hexokinase (1st enzyme in glycolysis) as well as phosphofructokinase and glycogen synthase) and inhibits glucose-6-phosphatase ((opposite direction of hexokinase, same reaction) gluconeogenesis).]
Insulin promotes fatty acid synthesis.
Once glycogen synthesis has maxed out (i.e. about 30g, about 20% of the carbohydrate part of a studied meal, max in 4-6hrs,) then fatty acid synthesis IN THE LIVER takes over. Glucose is converted to free fatty acids (FFAs) and dumped back into the blood as lipoproteins-- which are then broken up into FFAs.
FFAs go into the adipose cells of the body. Glucose also goes into adipose cells-- via GLUT-- and are converted into glycerols. Glycerol+FFAs= triglycerides.
Thus, insulin's role is to store fat and/or oxidize glucose. Too little insulin will also trigger protein catabolism.
GLUT1 and GLUT3 account for 95% of the glucose transport to the brain. GLUT1 is for the blood brain barrier (the tight junctuions of the BBB are what require these channels), and GLUT3 is in the neurons. (pic here) GLUT1 is also found in muscle.
These are not insulin dependent (like GLUT4 is) so the brain can continue to get its energy. Not only does the distribution of GLUT 1 and 3 mirror capillary density and areas of relative glucose utilization, the GLUT1/3 densities can change depending on chronically increased (or decreased) need for glucose. Interestingly, nicotine, which increases brain glucose utilization, increases GLUT1/3 but not capillary density.
GLUT2 and 7 are in the liver. GLUT2 can also carry D-fructose.
GLUT5 is in the intestine, and some glial cells of the brain.
Type II diabetes is insulin resistance, not lack of insulin. There is not, at least initially, a problem with the pancreas's secretion of insulin in response to high glucose. The problem is at the level of the insulin receptor and/or GLUT, which become insensitive to the effects of insulin-- because there has been so much of it for so long. (For more info, see: News Physiol Sci. 2001 Apr;16:71-6.
Next up: how do antipsychotics affect glucose/insulin/transporters?
(For a review: What We Know About Facilitative Glucose Transporters )
Score: 3 (5 votes cast)
Modafinil vs. Clozaril
I'm researching modafinil for my last post, and bam-- I see this thing. How is no one talking about this article? Does no one read anymore?
A short intro to EEGs and antipsychotics.
Generally, antipsychotics increase power across all frequencies, but each drug (or receptor system) is associated with a specific frequency's power increase. Additionally, antipsychotics' effects are region specific: here, the prefrontal cortex gets the majority of the effect.
 and 1 mg kg-1 s.c. (B); chlorpromazine 0.5 mg kg-1 i.p. (C) and quetiapine 2.5 mg kg-1 s.c. (D) on EEG spectral power in rats.")
Effects of haloperidol 0.5 mg kg-1 s.c. (A) and 1 mg kg-1 s.c. (B); chlorpromazine 0.5 mg kg-1 i.p. (C) and quetiapine 2.5 mg kg-1 s.c. (D) on EEG spectral power in rats. Panels on the left show data from prefrontal cortex, panels on the right show data from sensorimotor cortex.
Everything is higher (i.e. above the line), but see how each drug or dose changes which frequency sees the most increase in power? So now you can make a comparison table:

Summarizing:
D2 blockers (haldol, racloperide): increase in 10-15Hz power band (lesser in 15-20Hz)
Pure 5HT2A blockers (MDL100907): increases 2 Hz power band
High 5HT2/D2 ratios (Risperdal, sertindole): peak synchronization at 7-10 Hz.
In other words, mixing receptors gets you a mixed effect on EEG.
Additionally, drugs with high alpha-1 blockade (Clozapine, chlorpromazine, quetiapine) increase in 8-10 Hz power band.
You would guess that mixing an antipsychotic with apomorphine (a dopamine agonist) might decrease these effects, and mostly this would be right. There seems to be more effect on the sensorimotor cortex, but this isn't today's message.
What do you think would happen if you mixed Provigil and racloperide (D2 blocker) or clozapine? With racloperide, Provigil increased the power in the 10Hz band in both prefrontal and sensorimotor cortex-- a lot.
But apomorphine added to clozapine had no effect-- and Provigil added to clozapine decreased-- and at high doses almost totally extinguished-- the effect. This is the opposite of what happens in a pure D2 blocker!

Effects of the co-administration of clozapine (CLZ, A) 0.2 mg kg-1 s.c. and modafinil (MOD) 62.5 mg kg-1 i.p. (B), 125 mg kg-1 i.p. (C) or 250 mg kg-1 i.p. (D) in rats. The ordinate represents the percentage change in EEG power. Vertical bars for each Hz show 95% confidence intervals. Panels on the left show data from prefrontal cortex, panels on the right show data from sensorimotor cortex.
The likely explanation is alpha-1 blockade. Clozapine is a potent alpha-1 blocker. The wakefulness promoting, and EEG, effects of Provigil, which has no affinity for adrenergic receptors, are strangely blocked by prazosin (an alpha-1 blocker.) Thus, the specific effects of clozapine on EEG synchronization must be through alpha-1, not dopamine (i.e. the opposite of racloperide), as evidenced by the fact that they can be negated by Provigil, but not by apomorphine (again, the opposite of racloperide.)
In other words, alpha-1 blockade is integral, not incidental, to the antipsychotic efficacy of clozaril. It's not just orthostasis. This bodes well for Seroquel as well. But the reason why alpha-1 blockade is important is not clear. More on this when I figure it out.
Score: 1 (1 votes cast)
Provigil vs. Cocaine
In an attempt to see if there is an interaction between cocaine and Provigil, 20mg or 40mg IV cocaine was given pre and post Provigil (400mg or 800mg) for 7 days. There was an interaction, but it turned out to be positive: Provigil reduced systemic cocaine exposure.
A safety study investigated (in 7 people) the interaction between cocaine (30mg IV) and Provigil (modafinil) 200mg or 400mg, or placebo, and found no synergistic effect on vital signs (T, BP, HR) or EKG. Not only did it not augment cocaine euphoria, it blunted it in one person.
In another study, 62 (mostly black) males addicted to cocaine were randomized to placebo, CBT, or Provigil 400mg. Abstinence, the primary outcome, was measured by benzoylecgonine in the urine. Patients on Provigil were abstinent longer, and produced fewer positive urines (i.e. fewer relapses.) Importantly, no one got addicted to Provigil.
Unlike cocaine and Ritalin (methylphenidate) Provigil did not produce "cocaine like discriminitive stimulus" (i.e. didn't feel like cocaine; Ritalin and cocaine do feel like cocaine.)
That's all we know about Provigil vs. cocaine so far, which is pitiful but not inconsequential. Given Provigil's near absence of terrible side effects, I say it's worth a try.
In the interest of completeness (and correctness) I have to correct the major paper, above (the 62 people with the urine tests) . The authors of that paper propose the following potential mechanism:
Its glutamate-enhancing action (Ferraro et al, 1998; 1999) might be clinically advantageous in cocaine dependence because the repeated administration of cocaine depletes extracellular glutamate levels
Except that the Ferraro paper doesn't actually say that. What it says is that it inhibits striatal and globus pallidus GABA, but doesn't directly affect glutamate. In order for it to have any effect on striatal glutamate, you needed 300mg/kg (i.e. 21,000mg. See you on the other side.) Given that GABA and glutamate are opposites (i.e. glutamate goes up because GABA goes down), it's probably a small point, but not an insignificant one: if it directly increases glutamate, it could antagonize Lamictal or even potentially cause seizures (and it does neither.)
The second Ferraro reference finds essentially the same thing: inhibition of medial preoptic area and posterior hypothalamus GABA, and consequently glutamate increases. And again, all of this occurs at preposterously high doses (100-300mg/kg.)
In interesting side finding of Ferraro's study is that the medial preoptic area and posterior hypothalamus are primarily controlled by tonic GABA inhibition; consequently modafinil's (or any drug's) effect of increasing glutamate in these areas can be blocked by giving a GABA-A antagonist.
So Provigil operates by (probably) by antagonizing GABA, not specifically by enhancing glutamate (neither synthesis of or transport of).
To further complicate this picture, it may be that the effects on GABA and glutamate are both indirect, and really the result of serotonin agonism. In an earlier study by the same guy, the decreases in GABA were partially prevented by a 5HT3 blocker (think Zofran, Remeron). Does Provigil work through serotonin? In a later study, the same guy finds that at 100mg/kg, Provigil does, after all, increase serotonin in the medial preoptic area and posterior hypothalamus. (At lower doses, 10-100mg/kg, it increases serotonin in the cortex, dorsal raphe and the amygdala.) (And in another study, (yes, by that same guy again,) 3mg/kg Provigil, which in itself has no effect on serotonin, synergistically augmented serotonin increase to fluoxetine and imipramine.)
We already know that Provigil can reduce the sedation that comes from varying drugs, like SSRIs, general anesthesia, haloperidol, and chlorpromazine. It would be interesting to see if Provigil was unable to improve sedation on Remeron, supporting the 5HT3 hypothesis.
Good luck out there.
Score: 0 (2 votes cast)
Are Antipsychotics Overprescribed In Kids?
According to USA Today, 2.5 million antipsychotic prescriptions a year are written for kids under 18. The rate for privately insured kids is 6.5 in 1000-- it has to be easily ten times that for Medicaid kids.
The FDA database has 45 deaths; 6 from diabetes, the rest from CV disease, liver failure, suicide, etc. There were 41 pediatric NMS cases.
According to the article, 13% of antipsychotic prescriptions are for bipolar disorder.
So are antipsychotics being overprescribed? The answer is yes, but not for the reasons cited in the article.
The article, indeed, all articles about pediatric psychiatry, make a special point about how these medicines are not FDA approved for kids. This is absolutely meaningless. FDA approval requires two double blind, placebo controlled studies. These studies are universally taken on by the drug companies. No drug company would ever assume the massive risk of such a study-- let a lone two-- in kids. How do you recruit the study subjects? What parent is going to allow it? Rich parents? No chance. So it will have to be Medicaid parents-- and thus will come the Tuskegee-like charges, dripping with the obvious social and racial implications of pharma testing on poor minorities. Pharma is already loathed; they're not going to take any risks for the sake of a medal from the FDA. So there will not be any new pediatric indications for psych meds. Not in this climate. Think this hurts Pharma? It's your kids that suffer.
But don't be confused by crypto-socialist hysterics who say that Pharma will do anything for a profit, including peddle drugs to kids. Drug companies do not market these antipsychotics for kids. They are paranoid to a fault about doing this; they know everyone is scrutinizing them, especially lawyers. If you are a child psychiatrist who sees no adults, reps cannot even call on you. And if they call on you for other things, they cannot mention the use in kids. In the past five years, it has never-- never-- happened that a rep detailed me about their use in kids.
The only two reasons these drugs are used in kids is because psychiatrists give them, and parents demand them.
First, the parents. They don't come looking for antipsychotics, specifically. But my experience is that they are unrealistic about what is going on with their kids; in near denial about the family dynamics impacting on the kid's behavior; and virtually devoid of insight into relatively obvious, though procedurally difficult, maneuvers that could improve the situation. If your kid doesn't sleep enough, and consistently-- if your five year old doesn't nap-- you cannot tell me your kid has ADHD. Period. Parents demand a diagnosis of bipolar disorder for their kids because it means the divorce had nothing to do with it. They demand another medication when the first one fails to get the kid to do math homework instead of playing Xbox all day. And their kids' marijuana and alcohol abuse can't possibly have anything to do with their own marijuana and alcohol abuse. Parents: don't flame me. Your situation is different, I know. I know.
Second, psychiatrists prescribe them because of the pressure to do something, in the face of consistent failure. They don't start with antispychotics-- they end up with them. They prescribe them out of desperation. This is why, in every story about a child getting sick from one of these medicines, they are, in fact, on several medicines. First they start with Ritalin. If Ritalin doesn't help, or there is a side effect, or they can't sleep-- then a second drug is added. Maybe this helps, but after a while something else happens-- and another drug is added to this. That's why psychiatry's current obssession with the detection of underdiagnosed "bipolar disorder" is so important. This diagnosis justifies, and encourages, polypharmacy.
It is psychiatry's ridiculously dangerous, and ultimately doomed, paradigm: if you are not doing well on a medication, you must be so sick that you require two medications. It seems to have occurred to no one in psychiatry that failure on a medication could mean that it was the wrong medication.
The reason this polypharmacy madness is even possible is psychiatry's obsession with diagnosis, labels-- with semiotics.
What makes a drug an antipsychotic? Well, it treats psychosis. Fine-- but does that exclude its efficacy for something else? If it is later found to be efficacious in, say, depression, then what do you call it? Is the drug an antipsychotic that's also good for depression, or an antidepressant that's also good for psychosis?
There's no value in the label "antipsychotic" or "antidepressant" except what we give it. It's a drug that treats psychosis and depression, not an antipsychotic that treats depression (or the other way around). If you can't see the difference, stop reading now and go back to watching American Idol.
For example, why are antipsychotics viewed as "off label" for kids? The word "antipsychotic" is meaningless. Antipsychotics are tested against a scale, like the Brief Psychatric Rating Scale. But these scales measure a lot of things, like depression, and not just psychosis.
And at what point did we start making a distinction between psychosis and "dementia related psychosis?" Or bipolar depression and regular depression? Why do we need separate FDA approvals? Does someone know something about the physiology of these disorders that I don't? Do we need to start approvals for "diabetes related depression?"
Saying an antipsychotic is worse than an antidepressant for depression is a valueless statement, especially in the absence of data on this question. You are actually better off asking, "which is better for depression, blocking the serotonin transporter or blocking 5HT2a receptors?" See? Put this way the distinction seems less obvious. And even that question is valueless, as there is nothing (that we know of at this time) that allows us to say what effect either pharmacologic maneuver actually has. 5HT2A blockade does what again? Really? Do you have any evidence for that at all? And no more post hoc ergo propter hoc nonsense. David Hume laughs at you.
A Simpson's reference is helpful here:
Homer: Not a bear in sight. The Bear Patrol must be working like a
charm.
Lisa: That's spacious reasoning, Dad.
Homer: Thank you, dear.
Lisa: By your logic I could claim that this rock keeps tigers away.
Homer: Oh, how does it work?
Lisa: It doesn't work.
Homer: Uh-huh.
Lisa: It's just a stupid rock.
Homer: Uh-huh.
Lisa: But I don't see any tigers around, do you?
[Homer thinks of this, then pulls out some money]
Homer: Lisa, I want to buy your rock.
I know. The FDA, the Scientologists, socialists, the parents at the end of their ropes- the easy thing to do is blame Pharma. I'm in the strange position of having to be a Pharma apologist, to be the only doctor willing to defend Pharma. There are plenty things I don't like about the way Pharma conducts business, but I can't voice these complaints because I have to use the time countering these inane attacks. I know what will happen if the Pharma critics get their way.
You think Pharma should have no sales contact with physicians? Fine. Now deal with the consequences.
Score: 3 (5 votes cast)
CATIE Reloaded
And enough with the notion that medication compliance is a good proxy for overall efficacy.
All of these horrible psychiatry studies-- CATIE, Lamictal and Depakote maintenance trials, etc-- keep telling us how long patients stay on medications, because they say this means the drugs are working. The authors think that if a drug is working, they patient will stay on it. But you would think this only if you didn't actually treat many patients. I can make a similar argument that staying on a medication is inversely related to efficacy-- because when a patient feels better, they simply stop taking their meds.
Think about antibiotics. People don't finish the full 14 day course, precisely because they feel well. If they felt sick, they would probably take them longer than 14 days. In fact, people overuse these antibiotics even when its a virus, despite the antibiotic having no efficacy at all. They will demand an antibiotic even though know that it shouldn't be doing anything.
Same with pain meds. Oh, that's an acute problem? How about the chronic problems of diabetes and hypertension. People will skip/miss/forget doses when they feel asymptomatic, and will be more compliant when they have symptoms associated with these illnesses (e.g. headache, dizziness, etc.)
Look, I'm not telling you that compliance and efficacy aren't related. I am saying that if you want to measure efficacy, don't use compliance as a proxy-- go measure actual efficacy. And don't tell me it's too hard. You got $67 million for this study. Find a way.
Score: 6 (6 votes cast)
CATIE: And Another Thing
Score: 3 (3 votes cast)
CATIE: Sigh
1. You know, if you're going to be rigorous about BID dosing schedules because the FDA requires it, why so liberal with total dosing for Zyprexa? A mean dose of Zyprexa is 20.8 is way (150%) above FDA guidelines. For comparison, that would have meant dosing Geodon at 240mg, Seroquel at 1000mg, and Risperdal at 6mg. BTW: a mean of 20.8mg means that a lot of people were dosed with MORE than 20.8mg (max=30mg).
2. The miracle here isn't that Zyprexa won, but that Zyprexa 20mg barely won against Geodon 114mg.
3. Why Trilafon (perphenazine)? Originally you thought all conventionals were the same; so why not Haldol? Or Mellaril? You say it's because it had lower rates of EPS and TD, which is fine, but then why exclude TD patients from that arm?
4. So you excluded patients with tardive dyskinesia from the perphenazine group (fine) but then had the nerve to say people tolerated it as well as other meds? Do you think maybe people who have TD may have different tolerances to meds? Different EPS? Different max doses? That they're just different?
5. You can't generalize from an obviously slanted "typical" arm to all other typicals. If you chose Trilafon over Haldol because of better tolerability a priori, you can't now say that "typicals" have equal tolerability to atypicals. Why not pick two typicals of differing potencies (like Mellaril and Haldol) and infer from there?
6. Do you actually believe-- does anyone believe-- that any of these patients are compliant with BID regimens? Especially with sedating meds like Seroquel?
The secret to understanding CATIE 2 is to understand that there are two CATIE 2s.
CATIE2-Efficacy: People who dropped out of CATIE 1 because their med didn't work were randomized to Clozail, Zyprexa, Risperdal or Seroquel. On average, new Clozaril switches stayed on 10 months, everyone else only 3. 44% of Clozaril stayed on for the whole 18 month study; only 18% of the others completed the study.
CATIE2-Tolerability: People who dropped out of CATIE 1 because of side effects (not efficacy) were randomized to Zyprexza, Risperdal, Seroquel, and Geodon (not Clozaril.) Risperdal patients stayed on for 7 months, Zyprexa for 6, Seroquel for 4 and Geodon for 3.
CATIE2-Efficacy is fair. If you fail a drug, you're likely to do better on Clozaril than anything else.
CATIE2-Tolerabilty makes no sense at all. The reason Geodon was used is because it has "very different" side effects. Hmm. How? "In particular, ziprasidone [Geodon] was known not to cause weight gain." But this assumes that the intolerability of the first antipsychotic was its weight gain.
Most importantly is this: if a patient couldn't tolerate their first antipsychotic, how likely is it that it was effective? In other words, if it wasn't tolerable, it wasn't efficacious-- these patients could have been in CATIE2-Effectiveness study. So how did they choose?
Easy: they gave the patient the choice: Geodon or Clozaril? Out of 1052, half left altogether. 99 went into the Clozaril study (CATIE2-Effectiveness) and 444 went into Geodon (CATIE2-Tolerability.) Of the 444 in the Tolerability trial, 41% were actually labeled first drug non-responders. 38% were labeled as not tolerating their first drug, but of those, who knows how many were also nonresponders?
And 74% dropped out again.
If you take the 444 in the Tolerability study and divide them into two groups:
- those who left CATIE1 because of lack of efficacy: then switching to Zyprexa or Risperdal kept them on their meds longer. (Which makes no sense again: this is the same thing as the CATIE2-Effectiveness, where (except for Clozaril) there was no difference between Seroquel, Zyprexa and Risperdal.)
- those who left CATIE 1 because of lack of tolerability, then it made no difference what you switched to.
Sigh.
And what's with the blinding? In every other study with a clozapine arm, you equalize the weekly blood draws by making everyone have to submit to them. But in this case, they unblinded clozapine so as not to have to subject all these people to blood sticks. Except they were subjecting them already-- they were checking blood levels.
And where was perphenazine? "[CATIE1] did not anticipate this unexpected result [that perphenazine would be as efficacious] that challenged the widely accepted (but never proven) belief that the newer atypical antipsychotic medications are better than all older antipsychotic medications" and so was not considered for CATIE2. Apart from the fact that it is simply untrue that anyone thought the atypicals were more efficacious than the typicals, it is furthermore untrue that that the authors did not "anticipate this unexpected result." In 2003, after basically doing Medline meta-analysis, they found that "not all of them were substantially different from conventionals such as perphenazine."
What's funny about these guys is how they conveniently lump all typicals together but arguing for differential effects of individual atypicals; then argue typicals are different from each other to justify picking Trilafon; and then say atypicals are different from each other ("not all of them were different") but typicals are all pretty much the same ("conventionals such as perphenazine.")
Bottom line:
The stated purpose of CATIE2 was to help clinicians decide which drug to switch to if patients a) failed their first drug; b) couldn't tolerate their first drug.
The divorce rate in America is 40-50%. Say you get divorced, and a friend says, I have two women for you, Jane and Mary. If the problem with your first wife was that she didn't turn you on, you should marry Jane. If the problem with your ex was that she was annoying, you should marry Mary.
What's going to happen here is that your second marriage, to either girl, is doomed. Certainly more than the national average of 50%. How long is it going to take before your second wife doesn't turn you on either? How long before you find stuff intolerable about her? The answer is, more likely than your first marriage-- say, 75%-- because the problem isn't your wives, it's you. You've framed the question in an idiotic and arbitrary manner. You don't get married to get turned on OR to be with someone who isn't annoying. You want the marriage to have both simultaneously, and much more. These things are not separable. This is CATIE2. A meaningless dichotomy-- efficacy and tolerability are not separate, let alone opposites-- used to create a false paradigm of medication selection.
Score: 3 (5 votes cast)
Clozaril: FDA Misses The Point, Again
As you may know, when prescribing Clozaril (clozapine), a complete blood count with differential (CBC w/ diff) has to be checked every two weeks, because of the risk of agranulocytosis.
The FDA has relaxed these requirements: now, you have to check weekly for the first six months; then every two weeks for six months, then only monthly after that. You have to show WBC >3500/ml, and ANC>2000/ml. (That's white blood count and absolute neutrophil count.)
Don't think for a millisecond this was done because the FDA did a rigorous re-evaluation of safety data. This is the FDA that black boxed antidepressants for suicide and antipsychotics-- oh, sorry, only atypical antipsychotics, even though typicals are as bad, if not worse-- for death in patients with dementia related psychosis.
What's stupid about this is that agranulocytosis is the least of anyone's problems. In the Clozaril National Database (1990-1994) (1), there were 99,502 patients. 382 (0.4%) got agranulocytosis, and 12 died (that's 0.001%). The number of clozaril related deaths (all kinds) was more than 400.
In an Italian study, the rates of neutropenia are about 0.9%, and agranulocytosis 0.7% (2)
They are, however, dying in not insignificant numbers by other things.
Consider a Maryland finding: of the 2046 clozaril patients from 1990-2000, three died of new onset diabetic ketpacidosis. (0.15%) None had had diabetes. (3) Or the Israeli study (4) that found that 4/561 clozaril patients had sudden death-- 10 years younger, healthier, and 4 times the rate of non-clozaril treated sudden deaths. NB: no one died from agranulocytosis.
How about myocarditis: 8000 patients over 6 years: 15 myocarditis, 8 cardiomyopathy; 6 died. That's 0.3% 5 of the 6 deaths occurred in first month (that's right: month). (2) Given the rapidity of death, the authors speculate it's an acute hypersensitivity reaction (i.e. IgE/Type I).
A review of Pubmed/MEDLINE from 1970-2004 found rates of fatal myocarditis/cardiomyopathy to be between 0.015% and 0.188%.
An oft cited article by Walker examined 67072 clozapine patiens from 1991-1993, and found that of the 396 deaths, the most common cause was pulmonary embolism. (FYI: Zornberg found that exposure to low potency antipsychotics massively increases the PE risk to (OR 24 for low potency, 3 for high potency; not dose related, usually occurred in first three months.)
In contrast to Hagg's finding of 12 cases of PE/DVT, and a frequency of about 0.03%, another study of 13000 inpatients over 6 years found 5 PEs, i.e. a rate of 0.038%; but this was no different than typical neuroleptics of non-treated.
Look, I'm not saying to ignore agranulocytosis. I'm saying that when your patient's heart explodes, you can't say, "but the FDA only said CBCs!" You need to be checking EKGs. And when the lawyer asks you how most people die when on clozapine, it'll look really bad when you give the wrong answer.
Score: 5 (5 votes cast)
Subtypes of OCD
CNS Spectr. 2006 Mar;11(3):179-86.
The authors examined the difference between two (theoretical) subtypes of OCD symptoms. Based on the earlier work of Lee and Kwon, obsessions can be divided into autogenous and reactive.
Autogenous: occur without identifiable or likely stimuli; repetitive; disturbing;
Examples: "sexual, aggressive, or immoral thoughts, images, or impulses." The sudden obsession to rape; or seeing a red shirt, which signifies raping.
Coping strategy: suppress these thoughts
Reactive: caused by identifiable external stimuli, including thoughts of contamination, asymmetry, loss; are realistic;
Coping strategy:reduce anxiety (e.g. wash hands.)
They found some interesting differences:
Autogenous obsessives were more likely to be male, and older (34) and have older ages of onset (27);
Reactive obsessives were more likely female (60/40), younger (27) and younger age of onset (19).
No difference in either group in education or marital status; nor were there any major psychiatric comorbidities.
But autogenous obsessives rarely (1%) dissociated; reactives did dissociate (10%).
A few points. That aggressive/sexual thoughts go with males is no surprise. But that they are found in the older people is interesting. If you can see how primitive thinking occurs in reactive OCD, it makes sense they would get their symptoms at a younger age. It is known that cleaning and checking obsessions are associated with an earlier age of onset.
The prevalence of dissociation in reactive patients- or the absence of it in autogenous patients-- had already been suspected. Checking and symmetry symptoms (associated here with reactive type) had already been found to be more commonly associated with dissociation. (Personal diversion: my own clinical experience with pedophiles supports this-- that those with sexual obsessions are fully conscious of them; obsess over them, are familiar with every nuance. They know to molecular detail what the child looks like, how it moves; and are totally aware of their own behavior at every step of the molestation, even when they pretend a quasi-dissociative experience as a defense. They wouldn't dissociate during the acts because, in effect, it defeats the purpose of committing the act.)
Additionally, in other studies, Lee and partners (Telch, Kwon, etc) found that autogenous obsessions were more associated with schizotypal personality than to other OCD symptoms themselves (while reactive obsessions had no association with schizotypal.) 1 The authors use this finding to support the idea that autogenous obsessions represent cognitive issues, while reactive represent behavioral ones; autogenous obsessives obsess; reactive obsessives are compulsives. This is further supported another study finding that on Rorshach, autogenous obsessives, like schizophrenics, have severe thought disorder, while reactive obsessives do not.2 The belief that merely thinking a thought could make it true (formally called the likelihood bias of though-action fusion) sounds like the magical thinking in schizotypal, and in fact TAF is seen in schizotypals 4 1. It would be interesting if it could be investigated in autogenous obsessives versus reactive obsessives separately, the hypothesis being that autogenous obsessives display likelihood TAF, while reactives do not.
Clinically, it may be fair to say that people with autogenous type obsessions share schizotypal features, and cognitive/perceptual distortions, while reactive obsessions go with OCPD features and compulsive behaviors. So, do antipsychotics help autogenous obsessives (and not (or better than) reactive obsessives?)
In line with this, there is some evidence that schizophrenics and OCDs share some neurodevelopmental pathology. For example, using fractal dimension and CSF volume, one could accurately categorize schizophrenics or OCD patients, vs controls, with 90% accuracy.3 It would be interesting to see if these obsessives, based on brain pathology alone, could be distinguished between autogenous and reactive types. I suspect the answer will be yes.
1. J Anxiety Disord. 2005;19(7):793-805. Epub 2005 Jan 5.
2. J Clin Psychol. 2005 Apr;61(4):401-13.
Score: 3 (3 votes cast)
Deja Vu
I had a patient with a chief complaint of deja vu, so I looked it up. (BTW: turned out to be undiagnosed dementia in my patient's case.)
Best article I found was Wild E. J Neurol. 2005 Jan;252(1):1-7. Summarized (and all below references come from Wild's paper):
Definition: "any subjectively inappropriate impression of familiarity of a present experience with an undefined past." (Neppe, 1983)
Wild explains that the definition here is importantly specific: "subjectively inappropriate" means that the patient understands the familiarity is impossible (i.e. this is not a delusion.) "Undefined past" is a non-existent past, and the patient never pinpoints it (because it never happened).
Some theories:
Wigan (1985), Jensen (1868) and Maudsley (1889): a "loss of synchronicity" between two hemispheres of brain, so that they are working "separately but synchronously." Jensen also suggests it is familiarity of one part of the experience generalized to the whole.
Gestalt psychology: object-affect entities. An experience causes an affect, which is identical to the affect assocaited with an unrelated event in the past. Your brain interprets it as a rememberance of the object (as opposed to the affect.)
De Nayer:"tape recorder hypothesis:" you are remembering the event and recollecting the event at the extact same time.
Freud: the situation is similar to a suppressed fantasy, so the fantasy activates as a wish to make improvements in the current situation, so in essence it is the wish to turn back time.
Biology:
Associations with temporal lobe epilepsy suggested that that was the relevant neuroanatomy, but some interesting experiments suggest that deja vu is associated with limbic structures (especially hippocampus and amygdala) and not the temporal neocortex itself. Bancaud (1994) tried to synthesize the available information and proposed that the perception is encoded in the temporal neocortex and remembered in the hippocampus; affective memory is supplied by the amygdala. Then, these relate back to the temporal neocortex as a daja vu. Thus, a situation, as it is experienced and "recorded" (see De Nayer) activates deeper memory structures.

All of this appears to be lateralized to the to the temporal lobe ipsilateral to the dominant hand (which is really the non-dominant hemisphere-- it crosses). In fact, the point of all this was to use the symptom of deja vu to predict where a seizure focus would be. (Unfortuantely, a PET scan study could not confirm this lateralization.)
Because of the prominent association of deja vu with autosomal dominant "partial epilepsy with auditory features" a genetic contribution is suggested. This gene is the LGI1/epitempin gene (10q24).
Assessment should rule out depersonalization or flashbacks. If deja vu is short or infrequent, it is probably normal; but if recurrent, prolonged, or associated with physical sensations, consider TLE. If associated with anxiety or depression, consider psychiatric causes.
Other articles:
A study of 24 epileptics receiving direct stereotactic electrical stimualtion of the brain found that stimualtion of the entorhinal cortex produced more deja vu than the amygdala or hippocampus, while the perirhinal cortex was associated with the recollection of memories.
A prospective study of TLE patients and their "auras" (called simple partial seizures, i.e. seizures with no loss of consciousness, and include deja vu, weird tastes, etc)) found that the SPSs, deja vu, a warm sensation, a cephalic sensation, taste hallucination, and a "strange" sensation predicted an abnormal amygdala ipsliateral to the seizure focus. Though fear was the most common for all TLE patients, the single best predictor of an abnormal amygdala was... deja vu. As described by Wild, this occurred most commonly on the right (i.e. ipsilateral to handedness, or nondominant for language.)
A study of 14 patients with varying frontal lobe damage were tested for a number of memopry parameters; those with incorrect "feeling-of-knowing" had damage in the right prefrontal cortex.
Although deja vu is supposedly distinct from psychosis, and not related to dopamine, there's a case report of a 39yo physician-patient who took amantadine and phenylpropanolamine to ward off the flu, and got intense deja vus. They stopped when he stopped the medications. The authors find other case reports finding the same.
An interesting case report (two cases, actually) found that, contrary to the popular understanding that the deja vu shares some similarity to an actual past event, on formal testing these two patients had recollections which were unlikely related to previous familiarity ("incorrectly recognised low frequency words."). They then created justifications, i.e. they confabulated the recollections.
As an aside, there are 85 articles in Pubmed with the words "deja vu all over again" in the titles. Almost none actually had anything to do with deja vu. That's creativity for you.
Score: 3 (5 votes cast)
Is Schizophrenia Really Bipolar Disorder?
The answer should be so obvious that there shouldn't be an article on it. But there it is.
Lake and Hurwitz, in Current Psychiatry, conclude that schizophrenia is really a subset of bipolar disorder.
The author's initial volley is (sentence 3):
The literature, including recent genetic data (1-6) marshals a persuasive argument that patients diagnosed with schizophrenia usually suffer from a psychotic bipolar disorder.
Well that's a pretty powerful assertion, supported by 6 different references. Except for one thing: none of the six references actually support that statement.
- Berrettini: finds that of the various regions of the genome connected to bipolar disorder in genome scans, two are also found in scans of schizophrenia. Regions that overlap-- not genes, or collections of genes, but entire chunks of chromosomes. He says there are (perhaps) shared genetic susceptibilities, not that they are the same disease.
- Belmaker: A review article. No new data.
- Pope: specificity of the schizophrenic diagnosis-- written in 1978.
- Lake and Hurwitz: says there's no such thing as schizoaffective disorder, which would be groundbreaking stuff if it weren't written by the same author as this article.
- Post: Review article talking about kindling in affective disorders. In 1992.
- DSM-IV. Seriously.
I'm game if you are: find the "persuasive argument" that these references "marshal" and then we have something to talk about. What makes all this so hard to fathom is not the movement to lump the two disorders together, but rather to lump them together under the more arbitrary, heuristic diagnosis. It is schizophrenia, not bipolar, that actually has physical pathology. Let's review:
Brain anatomical findings:
- white-gray matter volumes decreased in caudate, putamen and nucleus accumbens. 1
- deficits in the left superior temporal gyrus and the left medial temporal lobe.2 3
- moderate volume reduction in the left mediodorsal thalamic nucleus (but total number of neurons and density of neurons is about the same) 4
- reduced gray matter volume, reduced frontotemporal volume, and increased volume of CSF in venticles 5 and 1
Physical features:
- Larger skull base and larger lower lip 1
- Velo-cardio-facial syndrome (22q11 deletion) 2
- vertical elongation of the face 3
- high arched palate 4
Granted, these aren't great; but try to find anything like this for bipolar.
Clearly, the Kool-Aid is delicious because they want us to drink it, too.
In the final section, magnificently entitled "What is standard of care?" the authors pronounce:
Antidepressants appear to be contraindicated, even in psychotic bipolar depressed patients.14,15 We suggest that you taper and discontinue the initial antipsychotic when psychotic symptoms resolve.
Which is great, except it's not true. If the authors have some evidence that antidepressants actually increase the switch rate, I'd love to see it: but for sure references 14 and 15 aren't it. At least, could "contraindicated" be a tad overstated?
The last two sentences of the whole article:
The idea that “symptoms should be treated, not the diagnosis” is inaccurate and provides substandard care. When psychotic symptoms overwhelm and obscure bipolar symptoms, giving only antipsychotics is beyond standard of care.
No references given for these outrageous statements, but given the relevance of their previous references I guess it really doesn't matter. "Substandard care?" "Beyond the standard of care?" Really? I'll see you in court.
Score: 5 (5 votes cast)
Do Antidepressants Induce Mania?
No.
This myth is the result of imprecise use of language. Many2 3 4 5 6 have detected a temporal association between the use of tricyclic antidepressants and episodes of mania in patients with bipolar disorder, though no plausible mechanism has been suggested, much less found. It has been assumed that this association could be extended to all antidepressants despite their substantial chemical and pharmacological heterogeneity. However, such an association between mania and selective serotonin reuptake inhibitors has not been found. In fact, the studies 7 8 9 which have been done strongly indicate that the rate of mania with SSRIs is no different than the normal switch rate of bipolar disorder. A recent study10 11 of venlafaxine, sertraline and bupropion found a 13% 10 week switch rate into mania or hypomania (excludes symptoms lasting less than 7 days), and an 18% one year switch rate, but this study was confounded by the concomitant use of mood stabilizers of dubious efficacy (lamotrigine, gabapentin, topiramate) in 22% of the patients, and the lack of a placebo arm to establish the baseline switch rate. Because of the significant phenomenological heterogeneity of the patients with “bipolar disorder” in clinical trials, a placebo arm is vital in order to compare results in different studies.
It must be reiterated that these are studies of temporal associations between mania and antidepressants, and no causal link can be inferred. Despite this, many continue to discuss the question in terms of the induction of mania by antidepressants. This betrays a bias that is supported neither by data nor logic. For example, studies of novel antidepressants cite rates of “induced” mania, while studies of antiepileptics discuss “breakthrough” manias, automatically presupposing a difference in the mechanism of mania. Similarly, it is popularly assumed that bupropion is less likely to induce mania than SSRIs. This assumption is formalized in the Expert Consensus Guidelines 200012, where bupropion “was clearly rated as the antidepressant least likely to precipitate an episode of mania.” It is not immediately obvious why this was so clear, as there is virtually no evidence indicating this. One single study13 (N=19) found bupropion to be less likely to induce a switch than desipramine. The final results of Post, above, are still pending, but it appears evident that if one has decided to accept the myth that antidepressants induce mania, there is no reason to believe bupropion is any less likely to do this.
The accumulated data in the field strongly suggest that the real risk in the treatment of bipolar depression is ineffectiveness, not mania.
-------------
2 Prien RF. Klett CJ. Caffey EM Jr. Lithium carbonate and imipramine in prevention of affective episodes. A comparison in recurrent affective illness. Arch Gen Psych 29(3):420-5, 1973 Sep.
3 Prien RF. Kupfer DJ. Mansky PA. Small JG. Tuason VB. Voss CB. Johnson WE. Drug therapy in the prevention of recurrences in unipolar and bipolar affective disorders. Report of the NIMH Collaborative Study Group comparing lithium carbonate, imipramine, and a lithium carbonate-imipramine combination. Arch Gen Psych 41(11):1096-104, 1984 Nov.
4 Boerlin HL. Gitlin MJ. Zoellner LA. Hammen CL. Bipolar depression and antidepressant-induced mania: a naturalistic study. J Clinical Psychiatry. 59(7):374-9, 1998 Jul.
5 Wehr TA. Goodwin FK. Rapid cycling in manic-depressives induced by tricyclic antidepressants. Arch Gen Psychiatry. 36(5):555-9, 1979 May.
6 Jann MW. Bitar AH. Rao A. Lithium prophylaxis of tricyclic-antidepressant-induced mania in bipolar patients. Am J Psychiatry. 139(5):683-4, 1982 May.
7 Amsterdam JD. Garcia-Espana F. Fawcett J. Quitkin FM. Reimherr FW. Rosenbaum JF. Schweizer E. Beasley C. Efficacy and safety of fluoxetine in treating bipolar II major depressive episode. Journal of Clinical Psychopharmacology. 18(6):435-40, 1998 Dec.
8 Peet M. Induction of mania with selective serotonin re-uptake inhibitors and tricyclic antidepressants. British J Psychiatry. 164(4):549-50, 1994 Apr.
9 Nemeroff CB, Evans DL, Gyulai L, Sachs GS, Bowden CL, Gergel IP et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry 2001; 158:906-12.
10 Post RM. Altshuler LL. Frye MA. Suppes T. Rush AJ. Keck PE Jr. McElroy SL. Denicoff KD. Leverich GS. Kupka R. Nolen WA. Rate of switch in bipolar patients prospectively treated with second-generation antidepressants as augmentation to mood stabilizers. [Clinical Trial. Journal Article. Randomized Controlled Trial] Bipolar Disorders. 3(5):259-65, 2001 Oct.
11 Post RM. Leverich GS. Altshuler LL. Frye MA. Suppes TM. Keck PE Jr. McElroy SL. Kupka R. Nolen WA. Grunze H. Walden J. An overview of recent findings of the Stanley Foundation Bipolar Network (Part I). [Journal Article. Review. Review, Tutorial] Bipolar Disorders. 5(5):310-9, 2003 Oct.
12 Sachs GS. Printz DJ. Kahn DA. Carpenter D. Docherty JP. The Expert Consensus Guideline Series: Medication Treatment of Bipolar Disorder 2000. Postgraduate Medicine. Spec No:1-104, 2000 Apr.
13 Sachs GS. Lafer B. Stoll AL. Banov M. Thibault AB. Tohen M. Rosenbaum JF. A double-blind trial of bupropion versus desipramine for bipolar depression. J Clin Psych 55(9):391-3, 1994 Sep.
Score: 3 (7 votes cast)
For more articles check out the Archives Web page ››